Hace cincuenta años, en 1956, tres médicos e investigadores de la universidad de Zurich, Andrea Prader, Alexis Labhart y Heinrich Willi, describieron por primera vez lo que hoy es conocido por Síndrome de Prader-Labhart-Willi, o más comúnmente Prader-Willi (SPW). En Octubre de 2002 tuvo lugar en Zurich un Simposio Internacional que permitió a la comunidad científica compartir avances y tratar asuntos para investigaciones futuras. Los resultados de este Simposio se presentaron en el libro "Síndrome de Prader-Willi como modelo para la obesidad"
El estudio y la terapia de este síndrome minoritario han progresado lentamente en los últimos años. En este 50 Aniversario de la descripción clínica del SPW, queremos rendir, con estos incompleto artículos biográficos, nuestro pequeño homenaje a estos tres importantes pediatras, cuyos nombres han marcado nuestras vidas.
Así pues, nuestro humilde tributo consistirá en dotar de mayor significado esos apellidos, que a veces, parece que sean los de nuestros hijos e hijas: Prader, (Labhart) y Willi, de forma que no sólo estas palabras, si no también la historia de los hombres que las trajeron hasta nuestros hogares perdure en la memoria de todas las familias y de todos los niños, jóvenes y adultos "Prader".
Con el afecto del que escribe y en nombre de muchas familias, gracias Andrea, Alexis y Heinrich por vuestra entrega y vuestro trabajo como primeros estudiosos de esta poco conocida, hoy, alteración genética. Vuestras vidas y vuestro legado perdurarán en la memoria de todas las familias, que para bien y en ocasiones para mal, nos acordamos de vuestros nombres un día .... y otro también.
**** **** ****
El profesor doctor Andrea Prader nació el 23 de diciembre de 1919, en Samedan, cantón suizo de Grisones . Inició sus estudios de Medicina en 1939 en Lausana y Zurich, terminando la facultad de Medicina en 1945, a pesar de que fue llamado al cumplimiento del deber en la caballería de las Fuerzas Armadas Suizas durante la Segunda Guerra Mundial. Tras la formación de postgrado en anatomía (con Töndury en Zurich) y medicina interna (con Vanotti en Lausana, cuyos trabajos estimularon su interés en los errores congénitos del metabolismo) decidió especializarse en pediatría
Se casó con Silvia Schucany en 1946, quien fue su compañera, también intelectualmente hablando, durante casi 5 décadas. Tuvieron tres hijos, dos varones y una mujer. En 1947, empezó su carrera profesional como médico auxiliar en el Hospital Infantil Kinderspital de Zurich. Su carrera siguió avanzando, sólo interrumpida por su visita a los Estados Unidos donde conocío a Lawson Wilkins (1894-1963), uno de los "padres" de la endocrinología pediátrica, cuya amistad fue determinante en el interés y el amor que, A. Prader, mostró durante toda su vida hacia la endocrinología pediátrica. En Nueva York, fue discípulo de Emmet Holt (1895-1974) en el Hospital Bellevue.
Pronto destacó dentro del grupo de discípulos y más cercanos colaboradores del profesor doctor Guido Fanconi. (1892-1979) en el Hospital de Zurich. En 1951, fue Oberarzt (jefe de residentes), en 1957 Privatdozent (Catedrático auxiliar) y, en 1962, fue nombrado director del Hospital Kinderspital y sucedió a G. Fanconi como Jefe de Departamento, un cargo que mantuvo hasta que se retiró en 1986.
El profesor A. Prader era un hombre delgado, de mediana estatura, de voz clara, mente abierta y un gusto moderno. Gran aficionado al arte, sobre todo a la pintura expresionista y también, curiosamente, al circo, lo que puede sorprender a muchos. "Impresionaba por su rápida comprensión de nuevos conceptos, por su abrumador pensamiento analítico, su estricta disciplina intelectual, su perspicacia y lo preciso de sus formulaciones". Dr. Silvestre Frenk.

A veces podía dar la impresión de ser una persona seca y poco amistosa. Debido al hecho de ser miope y, a menudo, mentalmente preocupado en asuntos importantes, algunos colegas jóvenes podían quejarse de que no les saludaba o reconocía al cruzarse con ellos por los pasillos. "Nunca he conocido a nadie como él. Lo que realmente me impresionó sobre él fue, no sus cargos honoríficos, ni sus premios, si no el hecho de que siempre era objetivo y justo, y que nunca hizo promesas que no pudiera cumplir. Si el estudiante más joven quería preguntarle una cuestión sensible, él siempre conseguía tiempo para escucharle y responderle cuidadosamente. Si, por el otro lado, alguna persona "muy importante" decía algo que no tenía sentido, él se levantaba y se iba, sin esperar a perder más tiempo." Dr. Milo Zachmann (1936-2002, premio Andrea Prader en 1988)
Pionero en incorporar a la práctica clínica, el progreso de la tecnología y las nuevas posibilidades diagnósticas y terapéuticas, se adelantó a la mayoría de los hospitales pediátricos del mundo. Aunque él era, de corazón, un endocrino pediátrico, tuvo amplios conocimientos en todas las áreas de la pediatría, resultano difícil enumerar todos sus logros científicos y académicos.
Fue el primero en describir la hiperplasia suprarrenal del lípido (con Siebenmann en 1955), la descripción del SPLW (con Labhart y Willi en 1956), la intolerancia hereditaria a la fructosa (con Froesch y Labhart en 1957) y la deficiencia de pseudo-vitamina D hipocalcémica como un tipo genético de raquitismo (con Ruth Llling), hoy conocido como raquitismo dependiente de vitamina D tipo I.
Presentó, con sus colaboradores, informes de casos poco comunes de interés fisiológico general, que supusieron un gran apoyo al establecimiento de laboratorios modernos (donde se refinaron los radioinmunoanálisis, la cromotografía del gas y la espectrometría de masa). Más adelante, facilitó e introdujo métodos de diagnóstico adicionales a los programas existentes tales como screening para el hipotiroidismo y la hiperplasia suprarrenal congénita (con Llling y Torresani).
Su interés y trabajo científico, fue fundamental para el análisis moderno del crecimiento y la pubertad. Él y su colega y amigo Tanner, en Londres, fueron de los primeros en introducir precisión matemática en el análisis del crecimiento y desarrollo. A. Prader inició y dirigió el estudio de crecimiento longitudinal de Zurich y su trabajo es continuado, todavía hoy. Coordinado con cuatro investigaciones en Europa y dos en África, este meticuloso trabajo de décadas dio lugar al establecimiento de referencias, no sólo para peso y talla, sino para otros indicadores, como densidad ósea y volumen testicular, desde el nacimiento hasta la edad adulta. Siendo precisamente estas investigaciones las que dieron lugar al orquidómetro de Prader . También fue uno de los primeros en utilizar hormona de crecimiento humana en pacientes con insuficiencia pituitaria.
En los años 60 y 70, tiempo en que la pediatría amenazaba en desintegrarse en varias subespecialidades, él lideró su grupo con buen hacer, y mostró como las subespecialidades podrían integrarse y como la pediatría como conjunto podía ser mantenida. Mientras que la mayoría de los nuevos especialistas tienden a sobreestimar la importancia de su propio campo y no siendo inusual que hiciera algún diagnóstico que había pasado por alto el especialista, A. Prader mostró las proporciones adecuadas a sus estudiantes y residentes. Sus clases las preparaba con todo esmero y como excelente conferenciante, claro y brillante, dirigido por la razón y la lógica, sus conferencias eran siempre motivo de interés en todos los congresos, locales e internacionales, a los que fue invitado alrededor del mundo, alcanzando fama internacional. "De sus conferencias siempre se podía llevar uno a casa un mensaje claro y útil."
Nadie sabe cómo hizo para gestionar un gran hospital y, a la vez, mantenerse al día en prácticamente todos los aspectos relacionados con la pediatría. Pero lo hizo, y los niños, sus pacientes, se beneficiaron de su atención a "todo el niño". Aunque aborrecía la charla "psicológica" sin contenido, y creía sólo en aquello que era claro, lógico y razonable, entendió la importancia de la verdadera psicología y la compasión. Nunca habló mucho de esto con sus discípulos, pero éstos recibían el mensaje cuando le veían en la cabecera de un niño muy enfermo, y percibían como sus finas maneras y su voz suave tenía un efecto calmante y tranquilizador.
Fue elegido para ser miembro del British Royal College of Paediatrics, la German Akademie der Naturforscher Leopoldina y unas cuantas sociedades más. Recibió doctorados honorarios en las Universidades de Tokushima, Frankfurt, Lyon y Zaragoza (donde su discípulo Dr. Nicolás Ángel Ferrández Longás estableció el Centro Andrea Prader para el estudio del crecimiento), así como importantes premios como el Premio Otto Nägeli (uno de los honores más altos en la ciencia suiza, en raras ocasiones otorgados a clínicos), y la medalla Heubner en Alemania. Naturalmente, también fue presidente, y más tarde miembro honorífico, de la Sociedad Suiza de Pediatría.
Gracias a su iniciativa, se fundó la Sociedad Europea de Endocrinología Pediátrica (ESPE). El año 2002 marcó el 40 Aniversario de esta exitosa sociedad: empezó con una reunión informal de 15 o 20 amigos y se ha convertido en todo un evento con unos 2.000 participantes en la edición de Montreal en el año 2001 (junto con la Sociedad Americana Lawson Wilkins). El Premio Andrea Prader se otorga anualmente a un miembro la ESPE como reconocimiento al liderazgo e implicación en la endocrinología pediátrica, siendo una de las formas de mantener viva su memoria.
Aunque los apreciaba, nunca dio demasiada importancia a los reconocimientos otorgados, y no los hacía públicos. Cuando volvía de un viaje, a menudo no mencionaba que había recibido algún cargo honorífico o alguna otra distinción. Sus discípulos normalmente se enteraban de ello gracias a sus conocidos en el extranjero. Andrea Prader se retiró en 1986 a los 67 años y tras la muerte de su esposa en 1995, su energía física y sus intereses intelectuales empezaron a decaer, como si a causa de la triste pérdida, hubiera perdido el gusto y la curiosidad que caracterizó su vida.
Pasó sus últimos años en una residencia para ancianos y el 3 de junio del 2001, falleció en la ciudad de Zurich, "uno de los personajes epónimos culminantes de la endocrinología pediátrica, descubridor y primer descriptor de cuando menos cuatro genopatías, gran clínico, intensamente comprometido con la enseñanza y, entre otros tópicos, con el cabal conocimiento del fenómeno del crecimiento y desarrollo corporal", el profesor doctor Andrea Prader.
**** **** ****
El profesor y doctor Alexis Labhart nació el 4 de mayo de 1916 en San Petersburgo, Rusia, el año anterior a la revolución. Hijo de padres suizos, en 1918 se trasladaron a Suiza y se instalaron en Basilea, donde A. Labhart realizó sus estudios. Se graduó en Medicina por la Universidad de Basilea en 1944, con una tesis sobre la tuberculosis en los campos de concentración.
Trabajó por un tiempo en un sanatorio en Davos y en 1947 entró como residente de Medicina Interna en el Hospital Universitario de Berna. En 1969 fue nombrado profesor de Medicina Interna de la Universidad de Zurich.
En su libro "Clínica de las Secreciones Internas" (1ª ed. Madrid: Morata, 1958) afirmaba que "…la única obesidad de condición endocrina en niños es la del Síndrome de Cushing, todas las demás formas de obesidad, tanto la del niño grueso con genitales al parecer pequeños, la lipodistrofia del tipo de calzones de montar, como la del Síndrome de Laurence-Moon-Biedl, no son provocadas por un exceso o un defecto de hormonas, ni por una composición hormonal falsa"
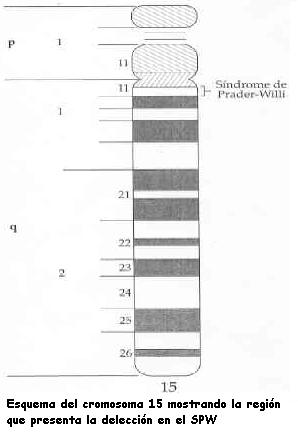
Con el transcurso del tiempo y por razones que desconocemos, el nombre de Labhart ha ido progresivamente desapareciendo al referirse al SPW, a pesar de su participación en la descripción del Síndrome de Prader-Labhart-Willi en 1956.
**** **** ****
Heinrich Willi nació el 3 de marzo de 1900 en Chur , Suiza. H. Willi estudió medicina en la Universidad de Zurich. Después de licenciarse en 1925, fue residente en el Instituto de Anatomía Patológica de Zurich y en el departamento de medicina del Hospital de Winterthur. Desde 1928 trabajó en el Hospital Infantil de Zurich, como miembro del equipo del profesor doctor G. Fanconi (1892-1979). Obtuvo su doctorado en Medicina en 1936 y al año siguiente sucedió al profesor Bernheim-Karrer, también fue director del "Säuglingsheim Rosenberg".
A parte de sus deberes académicos y hospitalarios, Heinrich Willi dirigió una gran clínica privada. Fue un innovador, desarrollando una gran labor en investigación médica a lo largo de su carrera, con importantes contribuciones en el campo de la Neonatología.
Se retiró en 1970 y a su muerte, el 16 de febrero de 1971 en Zurich, Guido Fanconi dijo: "Fue una ayuda amable y bendita para sus pequeños pacientes y sus temerosos padres. Lamentamos su muerte no sólo sus numerosos discípulos y sus antiguos profesores, sino también innumerables padres y antiguos pacientes, ahora también adultos. Todos lloran la muerte de una persona querida e inteligente que entendió como ser investigador, profesor y buen médico, al mismo tiempo."
**** **** ****
Quisieramos terminar este homenaje, con un fragmento del publicado por el Dr. Milo Zachmann, donde termina diciendo: "... era un privilegio para algunos de nosotros, a través de los años, cambiar gradualmente de colaborador joven, respetuoso y a veces muy aprensivo, a ser considerado su amigo. Entonces pudimos experimentar su humanidad, compasión, fino sentido del humor e ironía, y darnos cuenta que lo que parecía seco desde fuera era simplemente disciplina y - sí - la verdadera modestia que caracteriza a este honrado y famoso (...). Todos los que le conocimos más que superficialmente, y tuvimos el privilegio de aprender de él y trabajar con él, estaremos agradecidos de lo que nos dio, no sólo como nuestro profesor en pediatría y endocrinología, si no también como un ejemplo de continuidad, confiabilidad, austeridad y riqueza interior al mismo tiempo."
Después de una laboriosa búsqueda en la red, tenemos la impresión de poder hacer extensible a los Doctores Andrea Prader, Alexis Labhart y Heinrich Willi las palabras donde dice: "pasasteis infatigables por los bellos jardines de la medicina, la ciencia y el arte."
Por: Fernando Briones
Traducción: Mariona Nadal
Referencias:
Andrea Prader, in memoriam Dr. Silvestre Frenk
Instituto de Investigaciones Biomédicas, UNAM, Instituto Nacional de Pediatría, SSA, México.
Obituary Andrea Prader 1919 - 2001 Dr Milo Zachmann
Who named It?